
Learning the language of biology
How computers are learning biology and where this might go next.


As we’ve discussed in my first post, the merging of technology x biology is linked to the Danger Formula of Yuval Noah Harari (which is not necessarily a bad thing, but something we should be aware of). Two of the key components in the formula are biological knowledge and compute power. At a high level, the inclusion of these two components in the Danger Formula is obvious. But practically, this means that we need to get all of that biological knowledge into computer systems, into models that can perform the tasks we are interested in. In essence, we need numerical representations of biology that computers can process as an input, and we need those representations to be informative (to hold the biological knowledge).
Well, today we see more and more progress towards very informative representations for biology! Interestingly, this space of research was (in part) inspired by the huge progress in AI for natural language processing. In that way, the application of language models to biological data is a prime example of technology merging with biology. So in this blogpost, we’ll look into this space in detail and look at why language models for biology could become hugely important in the Tech x Bio space as a whole. Let’s start with some history!
The early history of models for biology
Arguably one of the first models for biology that has had a fundamental impact was Watson & Crick’s model for DNA structure. They discovered how nucleobases paired with one another (A-T and C-G) and form the three dimensional structure of DNA macromolecules. Their groundbreaking work in 1953 has been the foundation for molecular biology ever since. And that foundation was laid by empirical testing and evidence-based models.
But many things in biology are complex. As scientists began to research more complex systems, bottom-up approaches like Watson & Crick’s did no longer suffice. Rather, scientists began to look at things from a top-down perspective: observing the emergent properties of biological systems and figuring out the underlying rules to construct a model for that.
By 1978, Margaret Dayhoff had introduced the PAM matrices, which are matrices that capture the rate at which amino acids in protein sequences change (are replaced by one another) throughout evolution. PAM stands for Point Accepted Mutation and is a model for natural evolution. Margaret developed these matrices with a classic top-down approach: she observed the differences in closely related proteins and figured out the underlying rules (the rate at which amino acids are replaced) based on those observations. One downside is that Margaret looked at very closely related homologs of proteins (related from a common ancestor). Because of this, using her substitution matrices to study and align evolutionary divergent sequences did not work very well. Tackling this limitation, in 1992 Henikoff and Henikoff came up with their BLOSUM series of matrices (BLOck SUbstitution Matrix) that were constructed based on multiple alignments of evolutionary divergent protein sequences. An example of the most famous of such matrices is shown below (the BLOSUM62).

These substitution matrices are early examples of the top-down approach to biology in action. Moreover, some would argue that they are the first example of what we call ‘foundation models’ for biology. In short, a foundation model is a model that is trained on a large amount of data and is useful for a variety of downstream tasks. To some degree, substitution matrices indeed fit this description, although compared to today’s state-of-the-art, I would argue that we’ve made giant leaps of progress with modern techniques. The area where that’s most noticeable is the protein language space, which we’ll turn to next.
Protein language models
Looking at proteins is fascinating for multiple reasons, but a few years ago, scientists have started looking at protein sequences and their similarities to natural language (text, speech). They were intrigued by this comparison because of the significant progress in deep learning for understanding and generating natural language. Scientists reasoned that in fact, protein sequences share a lot of characteristics that are similar to natural language. Both text and protein sequences have local and global properties. In text, local properties might be the subject, the verb(s) and others, while in protein sequences these might be phosphorylation sites, local secondary structure elements and what not. Global properties in text refer to the topic or the language etc. In proteins, these might be the organism of which the protein comes from or its function, location in the cell and other properties.

On the other hand, proteins are not entirely similar to natural language.
First, we cannot read proteins. By simply looking at a protein (sequence), you won’t be able to say if it’s a human protein or a viral protein. While when looking at a text, you might be able to know what language it is, or recognize sentences and substructures through punctuation marks. This is more difficult in proteins.
Second, subtle differences in the sentence can have a big difference in meaning. Consider the sentence “I love you” versus “I loved you”. Even though those sentences only differ by one letter, their meaning is significantly different! In proteins, effects may be more aggregated (for example, hydrophilic chains in intermembrane sequences).
Third, protein sequence fold up into a 3D structure, which means that it’s not just the immediate surrounding that can have an effect, but also more distant interactions between amino acids take place.
Nonetheless, the space that naturally occurring proteins occupy should be learnable, if you reason from evolutionary pressures that encourage the reuse of components in proteins throughout evolution. Today, we see that this is indeed the case.
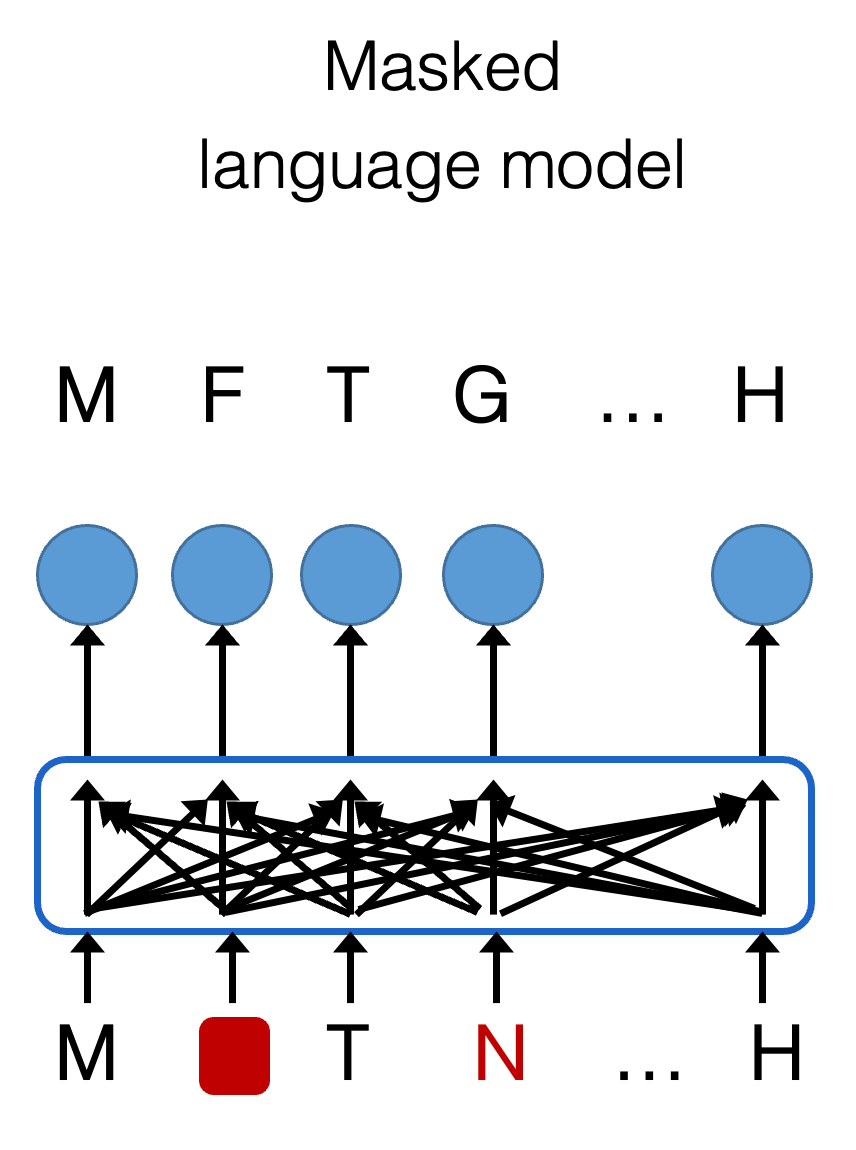
The current state-of-the-art in protein language models are a suit of models constructed by researchers at Meta AI, called ESM-2. We sometimes call these models ‘masked language models‘ because of the way they learn what protein sequences look like. In short, these models are trained by looping over every amino acid in the sequence and predicting the input sequence itself. Thus, the output is the same as the input, which is a concept we call self-supervision. Specifically, at each position, we ask the model to predict the amino acid in that place, given the context of amino acids around it. Crucially, what researchers do to enhance training is that some of the amino acids will be masked (hidden; red square in the figure below), and some others will be swapped for another amino acid (red amino acid in the figure below). Doing this, the model parameters are tuned by minimizing the errors that the model makes in predicting amino acids. In the end, we get to a trained model that is quite good at predicting what amino acids are in a certain place, given the context around it. We have trained a model that has ‘learned‘ what protein sequences look like. And that’s really the essence of these complex large language models!
The cutting edge and beyond
Beyond protein language models, today we’re also seeing progress towards genome-scale language models. It’s definitely interesting to try and extrapolate language models from individual proteins to full genomes or even proteomes. I can imagine that in various contexts, having a holistic representation of an organism (e.g. the SARS-CoV-2 virus) will be critical for downstream predictive tasks.
Even beyond academics, big companies are jumping into the biological language space. I’ve already mentioned Meta’s state-of-the-art ESM-2 model. Meanwhile, NVIDIA has launched its BioNeMo platform for drug discovery, powered by various of these biological language models. Salesforce is another notable company that is working in the protein language space, specifically focussing on protein generation.
Looking ahead, I speculate that this field will go in the same direction as the natural language space, towards large language models that are considered ‘foundation models’, handling various forms of input data (multi-modal) and performing well on various downstream tasks (multi-task). A deep dive into how this could unfold is for another time, but we can definitely conclude that progress in this space is happening fast and it’s only a matter of time before we see these models have actual impact beyond just research.